









Article provenant du magazine Semper – édition juin 2021 – www.dsb.lu
REGULATORY : Une rubrique originale de Semper Luxembourg en collaboration avec la Division de la Pharmacie et des Médicaments, sous la Direction du Dr Anna Chioti
Pour rappel, le dossier de la demande d’autorisation de mise sur le marché (AMM) comporte plusieurs parties dont la structure est harmonisée au niveau international pour faciliter la compilation des données et leur évaluation par les autorités. Ces trois parties techniques reprises dans la figure ci-dessous sont accompagnées d’éléments d’aides à l’utilisation du médicament par les médecins et les patients que sont le Résumé des Caractéristiques du Produit (RCP), la notice patient et les informations d’étiquetage.
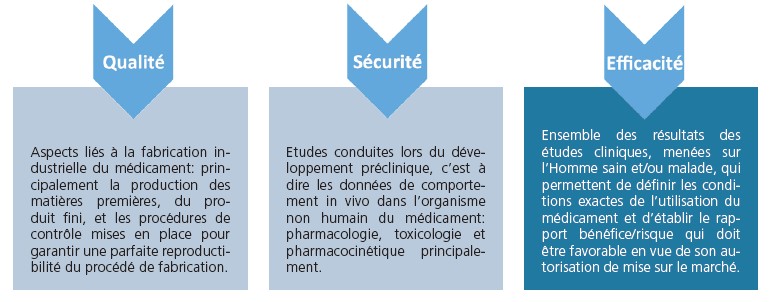
De toute évidence, un médicament (ou tout traitement médical) ne doit être utilisé que s’il apporte un bénéfice au patient. Le bénéfice prend en compte à la fois la capacité du médicament à produire le résultat désiré (efficacité) et le type et la probabilité des effets indésirables (sécurité).
Dans le cadre de l’évaluation par les agences des médicaments, l’efficacité est la capacité à produire un effet thérapeutique (p. ex., abaisser la tension artérielle). L’efficacité ne peut être évaluée avec précision que dans des conditions idéales (c’est-à-dire, lorsque les patients sont sélectionnés par des critères appropriés et se conforment strictement à la posologie). Ainsi, l’efficacité est mesurée sous le contrôle d’experts dans un groupe de patients les plus susceptibles d’avoir une réponse à un médicament, p. ex., dans un essai clinique contrôlé.
L’efficacité fait référence à la capacité d’un médicament à apporter un effet bénéfique (un rapport bénéfice/risque positif) lors de son examen au cours d’un essai clinique. La différence entre l’efficacité et l’efficience repose sur le fait que cette dernière fait référence au bon fonctionnement d’un traitement dans la pratique médicale en situation réelle, alors que l’efficacité mesure quant à elle le bon fonctionnement d’un traitement lors d’essais cliniques ou d’études de laboratoire.
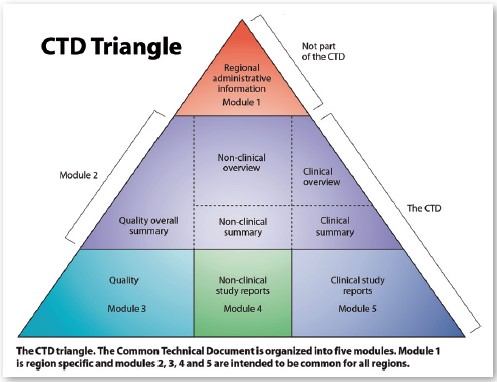
L’ensemble des données de qualité ainsi que les données de sécurité et d’efficacité précliniques et cliniques, sont organisées dans le Common Technical Document (CTD) qui est un format de dossier servant à la soumis¬sion des demandes d’autorisation de mise sur le marché (AMM). Comme son nom l’indique («document technique commun»), le CTD a pour principal intérêt d’être commun à la plupart des autorités de santé dans le monde (en tout cas les plus importantes: Europe, USA et Japon) pour la soumission d’un dossier de demande d’AMM. Le CTD a été mis au point par l’Agence européenne du médicament (EMA), son équivalent américain la FDA (Food and Drug Administration) et japonais (le ministère de la santé, du travail et du bien-être). Il est géré par l’ICH: International Conference of Harmonization, qui harmonise certaines parties de la réglementation des médicaments (pour plus d’informations sur l’ICH, vous pouvez consulter l’article précédent consacré la Qualité, Semper Avril 2021).
• Le module 1 est administratif et ne fait pas vraiment partie du CTD car il est spécifique à chaque région (Europe, USA,...).
• Le module 2 regroupe les Résumés des modules 3, 4 et 5.
• Le module 3 est le module Qualité. On y trouve le procédé de fabrica¬tion de la substance active et le procédé de fabrication du produit fini (= le médicament).
• Le module 4 contient les informations non-cliniques (ou pré-cliniques), c’est-à-dire les informations recueillies lors de l’usage du médicament chez l’animal.
• Le module 5 contient les informa¬tions cliniques, c’est-à-dire les informations recueillies lors de l’usage du médicament chez l’homme. Cela concerne principalement les don¬nées des études cliniques, mais aussi après commercialisation les données de pharmacovigilance avec les PSUR (= Periodic safety update report; c’est un document qui rassemble et analyse les effets indésirables provoqués par un médicament).
On ne le répètera jamais assez, mais ce n’est qu’après les multiples étapes du développement préclinique que les premiers essais thérapeutiques sur l’homme peuvent être réalisés. On parle alors de développement clinique. Les essais cliniques (ou essais thérapeutiques) sont une étape obligatoire et systématique du développement d’un médicament. Ils permettent de préciser l’effet d’un traitement chez l’homme, d’en déterminer l’efficacité ainsi que les éventuels effets indésirables.
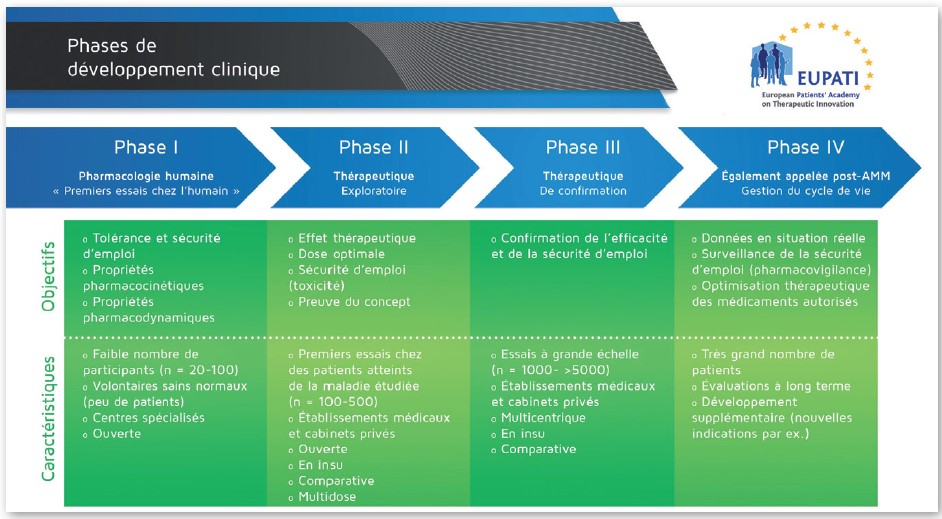
On distingue 4 phases dans les essais cliniques (voir illustration ci-dessous):
• Phase I: les essais sont, généralement, réalisés chez le volontaire sain (c’est-à-dire non malade) et ont deux objectifs majeurs. D’une part, s’assurer que les résultats concernant la toxicité, obtenus lors du développement préclinique, sont compa¬rables à ceux obtenus chez l’homme (quelle est la dose maximale du médicament en développement tolérée chez l’homme). D’autre part, il s’agit de mesurer, via des études de pharmacocinétique, le devenir du médicament au sein de l’organisme en fonction de son mode d’administration (absorption, diffusion, métabolisme et excrétion).
• Phase II: visent à déterminer la posologie optimale du produit en termes d’efficacité et de tolérance sur une population limitée et homogène de patients (quelques centaines). Les interactions médicamenteuses ainsi que la pharmacocinétique font parfois l’objet d’études dès cette phase.
• Phase III: ces essais, de plus grande envergure, sont conduits sur plusieurs milliers de patients représentatifs de la population de malades à laquelle le traitement est destiné. Il s’agit d’essais comparatifs au cours desquels le médicament en développement est comparé à un traitement efficace déjà commercialisé ou, dans certains cas, à un placebo, c’est-à-dire un traitement sans activité pharmacologique. Cette comparaison se fait, le plus souvent, en double insu et avec tirage au sort, c’est-à-dire que les traitements sont attribués de manière aléatoire sans que le patient et le médecin chargé du suivi soient informés de quelle attribution ils ont fait l’objet. Ces essais visent à démontrer l’intérêt thérapeutique du médicament et à en évaluer son rapport bénéfice/risque. C’est à l’issue de la phase III que les résultats peuvent être soumis aux Autorités Européennes de Santé dont l’EMA pour l’obtention de l’autorisation de mise sur le marché (AMM).
• Phase IV: ces essais sont réalisés une fois le médicament commercialisé, sur un nombre de patients souvent très important (jusqu’à plusieurs dizaines de milliers de personnes). Ils permettent d’approfondir la connais¬sance du médicament dans les conditions réelles d’utilisation et d’évaluer à grande échelle sa tolérance. La pharmacovigilance permet ainsi de détecter des effets indésirables très rares qui n’ont pu être mis en évidence lors des autres phases d’essai.
Pour en savoir plus, vous pouvez consulter les excellentes ressources développées par EUPATI: www.toolbox.eupati.eu/
.jpg)
Les travaux menés par l’ICH sous la rubrique «Efficacité» concernent la conception, la conduite, la sécurité et le reporting des essais cliniques. Il couvre également les nouveaux types de médicaments dérivés de procé¬dés biotechnologiques et l’utilisation de techniques pharmacogénétiques / pharmacogénomiques pour produire des médicaments mieux ciblés. Parmi les 20 guidances disponibles sur le site de l’ICH, on peut retenir le E6 relatif aux Bonnes pratiques cliniques (BPC), qui décrit les responsabilités et les at¬tentes de toutes les parties prenantes dans la conduite d’essais cliniques.
Le guide BPC couvre également les aspects de la surveillance, de la noti¬fication et de l’archivage des essais. Il est complété par des addenda concer¬nant les exigences pour les documents essentiels et les brochures des investi¬gateurs cliniques.
Initialement adopté en 1996, puis révi¬sé en 2016, il fait depuis 2019 l’objet d’un processus de «rénovation» en vue d’adapter les principes BPC aux types d’essais de plus en plus divers et aux sources de données utilisées pour sou¬tenir la réglementation et la prise de décisions en matière de soins de san¬té sur les médicaments, et offrir une flexibilité, le cas échéant, pour faciliter l’utilisation des innovations technolo¬giques dans les essais cliniques. Plus d’informations sont disponibles sur le site de l’ICH (www.ich.org/page/ef¬ficacy-guidelines#6-2).
Pour déterminer si un médicament ou un vaccin est efficace contre une maladie particulière, ou si un vaccin ou un médicament provoque des effets secondaires, nous utilisons le concept de preuve.
Toute recherche commence par la formulation d’une hypothèse. Une hypothèse est une idée suggérée comme explication de quelque chose. Si un grand nombre d’études de haute qualité confirment indépendamment une hypothèse ou des parties de celle-ci, nous la désignons comme une preuve. La preuve est la justification d’une hypothèse basée sur la recherche. Les preuves changent avec le temps à mesure que de nouvelles connaissances émergent et sont basées sur les meilleures connaissances et les plus à jour possible. Les preuves et l’expérience sont également deux choses différentes. L’expérience personnelle ou des observations uniques constituent des preuves faibles. Mais elles peuvent être utilisées pour formuler de nouvelles hypothèses qui peuvent être étudiées scientifiquement.
Lorsqu’ils testent une hypothèse, les chercheurs disposent de nombreuses méthodes de recherche différentes. La méthode la plus appropriée dépend du sujet à étudier. Il est également pris en compte si plusieurs (et bonnes) études aboutissent au même résultat. Si plusieurs études différentes menées par différents chercheurs aboutissent au même résultat ou pointent dans la même direction, les preuves sont solides et l’hypothèse est étayée. Il se pourrait également que la majorité d’un certain nombre d’études pointent dans une direction, tandis que quelques autres études pointent dans une autre direction. Cependant, des preuves solides dépendent d’une conception et d’études de recherche systématiquement, minutieusement et objectivement sélectionnées.
Les résultats individuels et les études constituent les pièces d’un grand puzzle. Lors de l’évaluation des études et du niveau de preuve, on considère si l’hypothèse sous-jacente est pertinente, comment l’étude est conçue, combien de personnes ont participé à l’étude, combien de temps a duré l’étude, quelle était l’ampleur de l’effet mesurable, quelles autres connaissances sont disponibles dans le domaine et, enfin, combien d’études vont dans le même sens.
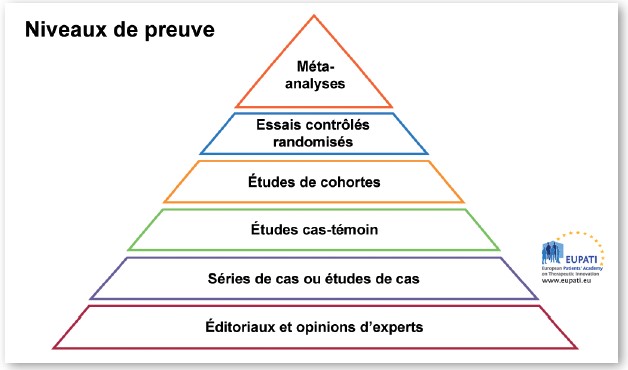
La robustesse de l’étude est également importante. La pyramide des preuves (ou hiérarchie des preuves) est utilisée dans la recherche scientifique en santé pour indiquer les types d’essais qui fournissent les preuves les plus solides. Les études avec la position la plus élevée dans la pyramide sont considérées comme les plus importantes.
Les essais randomisés, s’ils sont faits sur un nombre suffisant de patients, sont considérés comme ayant le plus haut niveau de preuve en matière thérapeutique.
«La randomisation a pour but de minimiser les biais et de s’assurer que les patients dans chaque groupe de traitement soient aussi proches que possible dans tous les aspects connus et inconnus. Cela garantit que toutes les différences trouvées entre les groupes concernant le(s) critère(s) utiles soient dues à des différences quant à l’effet du traitement et non à des différences entre les patients recevant chacun des traitements. Elle élimine le risque qu’un clinicien assigne consciemment ou inconsciemment un traitement à un type particulier de patient et l’autre traitement à un autre type ou qu’un certain type de patient choisisse un traitement tandis qu’un autre type choisirait l’autre.»
La «différence significative» peut avoir plusieurs sens. Elle peut tout d’abord correspondre à une différence qui est en fait importante pour le patient. Cependant, lorsque les auteurs des rapports de recherche affirment qu’il existe une «différence significative», ils font souvent référence à une «différence statistique.» Il faut savoir que des «différences statistiquement significatives» ne sont pas nécessairement «significatives» au sens clinique du terme. Une différence entre les traitements qui est peu susceptible d’être due au hasard (une «différence statistiquement significative») peut en pratique avoir peu d’importance ou n’en avoir aucune.
Donc la mention «statistiquement significatif» indique si la différence entre un médicament et un placebo, par exemple, pourrait n’être due qu’au hasard. Cela signifie qu’il est improbable qu’une différence aussi importante que celle observée ait pu être provoquée uniquement par le hasard. Les statisticiens utilisent des niveaux standard d’«improbabilité». Généralement, ils utilisent le terme significatif pour le niveau de 5% (parfois écrit p = 0,05). Dans ce cas, on dit qu’une différence est «significative», car elle a une probabilité inférieure à 1 sur 20 de survenir si tout repose uniquement sur le hasard.»
Une étude unique fournit rarement assez de preuves pour orienter des choix de traitement en matière de soins de santé. C’est pourquoi, les évaluations des avantages respectifs de traitements alternatifs doivent se fonder sur des revues systématiques de toutes les preuves fiables pertinentes. Les essais contrôlés de traitements impliquent d’examiner systématiquement toutes les preuves fiables pertinentes afin de déterminer ce qui est déjà connu, que ce soit par les recherches chez l’animal ou d’autres recherches de laboratoire, par l’intermédiaire des volontaires sains chez lesquels de nouveaux médicaments sont parfois testés ou par les recherches antérieures portant sur des patients.
Si cette étape est ignorée ou réalisée de façon incorrecte, les conséquences peuvent être graves, les patients en général, ainsi que les participants aux recherches, peuvent souffrir et parfois mourir inutilement et de précieuses ressources, tant pour les soins de san¬té que pour la recherche, seront gaspillées.
Pour en savoir plus et accéder à des formations www.training.cochrane.org/essentials

for articles/videos/studies

The articles can be sorted by therapeutic area or disease, but may also deal with more general topics not specifically related to a disease. These articles can be sorted as "other".
