









Regulatory: Une rubrique originale de Semper Luxembourg en collaboration avec la Division de la Pharmacie et des Médicaments, sous la Direction du Dr Anna Chioti
Cet article provient du magazine SEMPER – édition mai 2021 – www.dsb.lu
Dans le cadre des demandes d’autorisation de mise sur le marché (AMM) des médicaments par les firmes titulaires, les autorités compétentes exigent le dépôt de dossiers constitués de plusieurs parties dont la structure est harmonisée au niveau international pour faciliter la compilation des données et leur évaluation. Ces parties sont essentiellement consacrées aux données de qualité, sécurité et efficacité des médicaments.
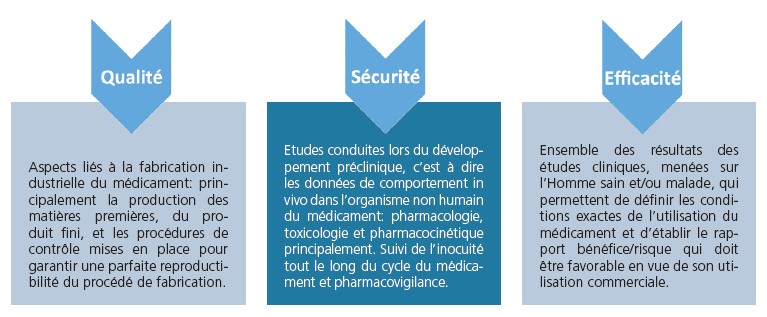
L’évaluation de la sécurité des médicaments est essentielle et ce tout au long de leur cycle de vie, depuis la découverte et le développement en laboratoire, jusqu’aux études cliniques chez l’homme en vue de l’obtention de l’autorisation de mise sur le marché (AMM). Mais les médicaments restent sous surveillance continue une fois commercialisés. Ainsi, le rapport bénéfices/risques des produits est évalué en permanence pour prendre notamment la mesure des effets indésirables connus ou nouvellement identifiés. En cas de risque pour la santé, un médicament peut se voir appliquer une restriction ou une modification des indications. Le médicament peut également faire l’objet d’un retrait du marché.
Depuis une quinzaine d’années, des plans de gestion des risques (PGR) sont mis en place et permettent de mieux connaître la sécurité d’emploi de certains médicaments, dès leur mise sur le marché, en les étudiant en situation réelle de consommation.
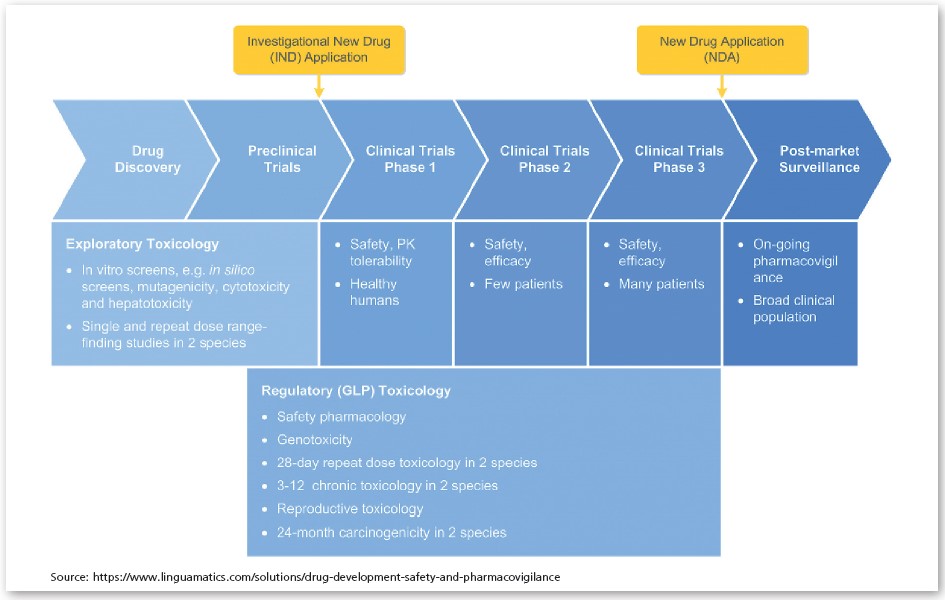
Le diagramme ci-dessous montre un aperçu des principales études d’évaluation de l’innocuité menées au cours du processus de développement du médicament.
La tragédie de la thalidomide au milieu du XXe siècle a déclenché une série d’activités qui faisaient partie d’un effort mondial visant à éviter une récidive.
La thalidomide a été synthétisée par la firme allemande Chemie Grunenthal en 1953. Après des tests sur des animaux et sur des humains, ayant démontré un bon profil de sécurité, la thalidomide a été lancée sur le marché Ouest-Allemand.
Dans les années soixante, la thalidomide était utilisée en Europe et au Canada chez les femmes enceintes comme sédatif et antiémétique. En 1961, Chemie Grunenthal a soumis l’autorisation de mise sur le marché auprès de la FDA aux Etats-Unis, qui réclamait alors des documents supplémentaires.
Les demandes de clarification émanaient de la survenue de cas de phocomélie dans divers pays européens ainsi que des cas de neuropathie périphérique. De plus en plus de mé¬decins ont été persuadés qu’il y avait un lien de causalité hautement probable entre la prise de thalidomide pendant la grossesse et la survenue de malformations congénitales.
La firme a dû ordonner le retrait de la molécule et celui de tous les autres médicaments contenant de la thalidomide au niveau mondial. Mais cette tragédie aura affecté environ 12.000 enfants dans 46 pays.
.jpg)
Désormais des résultats de tests spécifiques pour évaluer l’effet tératogène des nouvelles substances seront exigés par les instances qui approuvent les nouveaux médicaments.
En conséquence, à partir du milieu des années soixante-dix, la FDA a renforcé ses exigences, notamment en refusant de prendre en compte les résultats issus des essais cliniques réalisés dans d’autres pays, considérant qu’ils ne répondaient pas aux mêmes critères d’éthique et de sécurité pour les participants qu’aux Etats- Unis.
Au cours des cinquante dernières années, il y a eu une croissance constante de la science maintenant connue sous le nom de pharmacovigilance avec un virage exponentiel ces dernières années. Au cours de cette croissance, diverses terminologies et paramètres ont été introduits pour permettre la communication et les échanges entre les parties prenantes sur le terrain.
Par ailleurs, la portée de la pharmacovigilance a considérablement augmenté et est maintenant considérée comme incluant les domaines suivants:
• Effets indésirables ou réactions indésirables au médicament.
• Erreurs de médication.
• Médicaments contrefaits ou de qualité inférieure.
• Manque d’efficacité des médicaments.
• Mauvaise utilisation et/ou abus de médicaments.
• Interactions entre les médicaments.
• Expositions particulières: femmes enceintes ou allaitantes.
• Usages hors indications.
L’Agence européenne des médicaments (EMA) coordonne le système de pharmacovigilance de l’Union européenne (UE) et gère des services et des processus pour soutenir la pharmacovigilance dans l’UE.

Avant d’autoriser l’utilisation d’un médicament, les preuves de sa sécurité et de son efficacité se limitent aux résultats d’essais cliniques, où les patients sont soigneusement sélectionnés et suivis de très près dans des conditions contrôlées.
Cela signifie qu’au moment de l’autorisation d’un médicament, celui-ci a été testé sur un nombre relativement restreint de patients sélectionnés pendant une durée limitée.
Après autorisation, le médicament peut être utilisé chez un grand nombre de patients, pendant une longue période et avec d’autres médicaments. Certains effets secondaires peuvent apparaître dès lors dans ces circonstances.
Il est donc essentiel que la sécurité de tous les médicaments soit surveillée tout au long de leur utilisation dans la pratique des soins de santé.
Le droit de l’UE impose donc à chaque titulaire d’autorisation de mise sur le marché, à l’autorité nationale compétente et à l’EMA de mettre en place un système de pharmacovigilance.
Le système global de pharmacovigilance de l’UE fonctionne grâce à une coopération entre les États membres de l’UE, l’EMA et la Commission européenne. Dans certains États membres, des centres régionaux sont en place sous la coordination de l’autorité nationale compétente.
Le Comité d’évaluation des risques de pharmacovigilance (PRAC) de l’EMA est chargé d’évaluer et de surveiller la sécurité des médicaments à usage humain. Il est composé d’experts en sécurité des médicaments issus des autorités réglementaires des États membres, ainsi que d’experts scientifiques et de représentants de patients et de professionnels de santé désignés par la Commission européenne.
L’EMA soutient le PRAC en fournissant des données issues de la pratique clinique disponibles dans des dossiers de santé électroniques ou des bases de données de prescription.
Le Luxembourg est représenté au sein du PRAC par Anne-Cécile Vuillemin, Pharmacien Inspecteur, toxicologue, en charge de la pharmacovigilance au sein de la Division de la Pharmacie et des Médicaments (DPM).
L’Agence est chargée de développer et de maintenir EudraVigilance, un système de gestion et d’analyse des informations sur les effets indésirables suspectés des médicaments autorisés dans l’Espace économique européen (EEE).
EudraVigilance est un référentiel unique pour les rapports d’effets indésirables suspectés observés dans la pratique de la santé et les essais cliniques. Il est utilisé par les États membres, l’Agence et l’industrie.
Le PRAC évalue les signaux de sécu¬rité d’EudraVigilance et peut recommander une action réglementaire en conséquence.
L’EMA publie les données d’EudraVigilance dans la base de données européenne des rapports sur les effets indésirables suspectés des médicaments.
Les utilisateurs peuvent afficher le nombre total de rapports individuels d’effets indésirables suspectés soumis à EudraVigilance pour chaque médicament autorisé au niveau central.
| «Un PGR, c’est l’ensemble des dispositions mises en oeuvre pour minimiser les risques potentiels dans l’usage des médicaments.» |
L’Agence travaille en étroite collaboration avec un certain nombre de partenaires internes, notamment:
• La United States Food and Drug Administration (FDA): partage d’informations sur les questions de sécurité des médicaments et sur les mesures réglementaires prévues, l’information du public et la communication avant la prise de décision et la publication.
• L’Organisation mondiale de la santé (OMS): communication de toutes les mesures prises concernant les médicaments autorisés au niveau central qui peuvent avoir une incidence sur la protection de la santé publique dans les pays hors de l’UE.
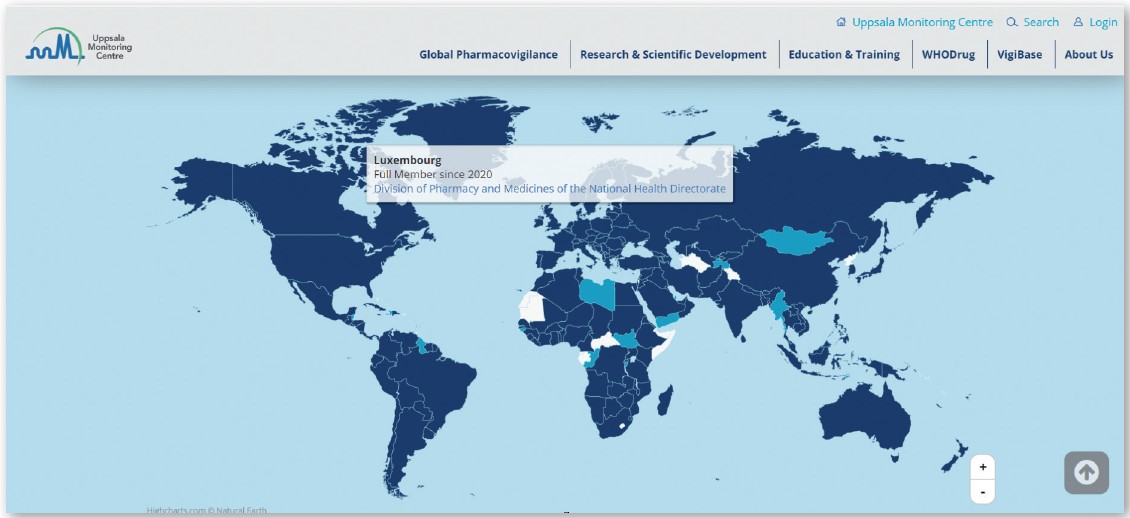
L’Uppsala Monitoring Center (UMC) a été créé à Uppsala, en Suède en 1978 en tant que Centre collaborateur de l’Organisation mondiale de la santé (OMS) pour la surveillance internationale des médicaments. L’UMC gère les aspects techniques et scientifiques du réseau mondial de pharmacovigilance de l’OMS.
La Division de la Pharmacie et des Médicaments a oeuvré pour que le Luxembourg rejoigne l’UMC en 2020.
Un PGR, c’est l’ensemble des dispo¬sitions mises en oeuvre pour minimiser les risques potentiels dans l’usage des médicaments. Avant la mise sur le marché, le PGR identifie les risques connus et potentiels. Après la mise sur le marché, le PGR fixe les moyens à mettre en oeuvre pour repérer les risques non prévus.
Le PGR est systématique pour tout nouveau médicament. En effet, toutes les spécialités pharmaceutiques mises sur le marché depuis 2005 en Europe, entraînent l’obligation, pour chaque laboratoire, d’assortir sa demande de mise sur le marché d’un Plan de Gestion des Risques (PGR). Ces PGR peuvent également comporter des mesures spécifiques additionnelles au niveau de chaque pays.
Le PGR n’implique pas qu’un médicament est spécialement «à risques», mais il constitue une garantie supplémentaire de suivi des événements de pharmacovigilance, qui feront l’objet d’une surveillance «personnalisée», en plus des procédures classiques de pharmacovigilance.
Le Plan de Gestion des Risques constitue un progrès en termes de surveil¬lance, car il implique d’évaluer de façon continue, dans les conditions réelles d’utilisation du médicament, le rapport bénéfice/risque de ce dernier (p.ex. essais cliniques post- AMM ou collecte de données dans des registres). Le PGR détaille également les actions concrètes mises en oeuvre pour minimiser les risques d’un produit (distribution de carnets de suivi, livrets d’information, programme d’accompagnement, etc.).

Depuis la crise COVID-19 et la nécessité de développer rapidement des vaccins sûrs et efficaces, la pharmacovigilance a eu un regain d’intérêt de la part des professionnels de la santé mais aussi de la part grand public. Les gros titres des journaux et les publications virales sur les réseaux sociaux d’effets indésirables graves et la pression politique qui peut en découler ne facilitent pas la tâche des professionnels de la pharmacovigilance.
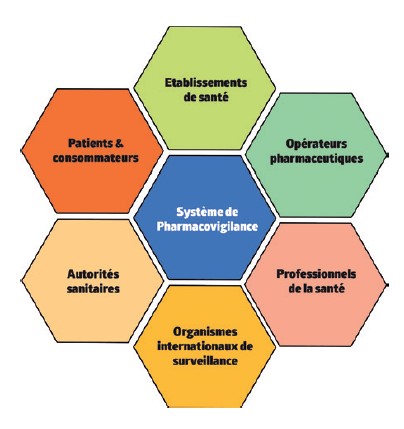
La surveillance continue de la sécurité des médicaments s’effectue en collaboration avec le CRPV de Nancy mais aussi avec, sur le terrain, les professionnels de santé et les patients. Les médecins, les médecins-dentistes, les pharmaciens et les sages-femmes ont notamment l’obligation de déclarer tout effet indésirable susceptible d’avoir été provoqué par un médicament et dont ils ont connaissance. Les autres professionnels de santé et les patients peuvent également déclarer les effets indésirables.
Ce système pour but de recueillir les informations utiles concernant les risques que présentent les médicaments pour la santé des patients et la santé publique de manière générale. Il permet de veiller à la sécurité d’emploi et au bon usage des médicaments, d’informer au mieux les patients et professionnels de santé et d’assurer en continu une balance bénéfice-risque positive pour les patients.
Ces informations concernent en particulier les effets indésirables survenant chez l’homme en cas:
• d’utilisation d’un médicament conformément aux termes de son autorisation de mise sur le marché;
• d’utilisation non conforme aux termes de l’autorisation de mise sur le marché;
• d’exposition professionnelle.
Elles concernent aussi certaines situations particulières avec ou sans effets indésirables associés telles que les erreurs médicamenteuses, les abus, les mésusages, les surdosages, les utilisations hors autorisation de mise sur le marché, les expositions accidentelles ou les expositions pendant la grossesse ou l’allaitement.
Ces informations font l’objet d’une évaluation scientifique, en vue no¬tamment de prévenir ou réduire les risques liés à la consommation de médicaments.
| «La pharmacovigilance est l’affaire de tous et le système de pharmacovigilance constitue l’axe principal d’un système de santé centré sur la sécurité du patient.» |
for articles/videos/studies

The articles can be sorted by therapeutic area or disease, but may also deal with more general topics not specifically related to a disease. These articles can be sorted as "other".
